Myokardisk iskemi og hjerteinfarkt: Cellulære forandringer, EKG og symptomer
Myokardiskemi: cellulære forandringer, EKG-tegn og utvikling av infarkt
I likhet med alle andre celler i menneskekroppen er kardiomyocyttene (hjertemuskelcellene) avhengige av ATP (adenosintrifosfat) som sin primære energikilde for å opprettholde homeostase og kontraktilitet. ATP produseres ved metabolisering av substrater som frie fettsyrer, glukose og i mindre grad laktat og ketoner. Under normale aerobe forhold dekkes hjertets energibehov hovedsakelig gjennom beta-oksidasjon av frie fettsyrer (ca. 60–90 %), mens glukoseoksidasjon spiller en sekundær rolle. ATP er drivstoffet for essensielle cellefunksjoner, inkludert membrantransport (ionepumper) og interaksjonen mellom aktin og myosin som muliggjør kontraksjon og relaksasjon.
Hjertet er et aerobt organ med liten kapasitet for anaerob metabolisme. Økt arbeidsbelastning, indusert ved økt hjertefrekvens, økt kontraktilitet (inotropi) eller økt veggstress (etterlast), medfører en lineær økning i myokardets oksygenforbruk (MVO2). Under fysiologiske forhold regulerer koronarsirkulasjonen blodgjennomstrømningen (koronar flow) gjennom autoregulering for å matche dette behovet nøyaktig. Dette innebærer at hjertet er avhengig av kontinuerlig perfusjon for å opprettholde både elektrisk stabilitet og mekanisk pumpefunksjon.
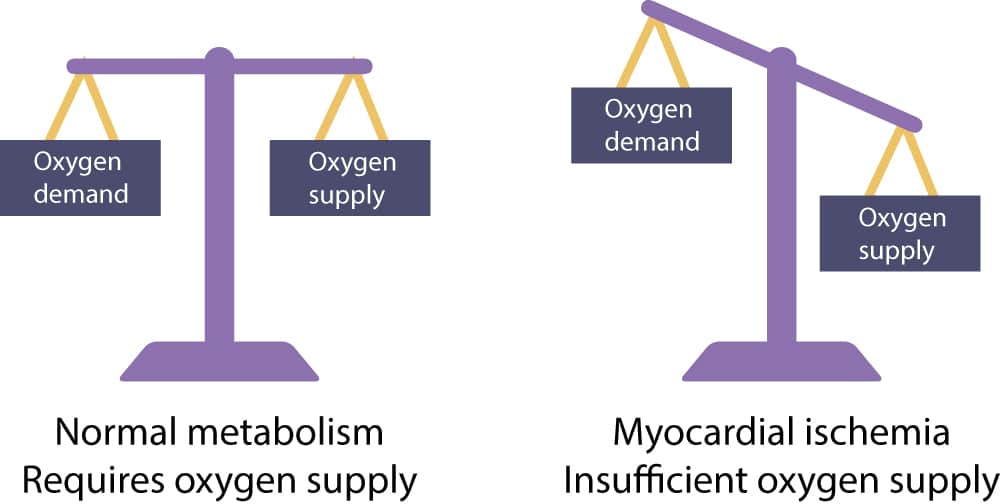
Myokardiskemi oppstår ved en ubalanse mellom oksygentilbud og oksygenetterspørsel (supply/demand mismatch). Når oksygentilførselen faller under det kritiske nivået som kreves for oksidativ fosforylering, reduseres ATP-produksjonen dramatisk. Dette utløser en umiddelbar metabolsk omstilling der myokardiet forsøker å kompensere ved å skifte til anaerob glykolyse. Selv om hjertet lagrer noe glykogen, er utbyttet av ATP fra anaerob glykolyse svært lavt sammenlignet med oksidativ metabolisme, og det medfører opphopning av laktat og H+-ioner (intracellulær acidose).
De cellulære konsekvensene inntreffer raskt:
- Nedsatt kontraktilitet: Allerede få sekunder etter okklusjon svikter kontraksjonsevnen («hypokinesi» eller «akinesi») i det iskemiske området. Dette er en beskyttelsesmekanisme for å spare ATP, men skyldes også direkte effekter av acidose på kalsiumhåndteringen i cellen.
- Elektrisk ustabilitet: Mangelen på ATP påvirker Na+/K+-ATPasen og andre ionepumper, noe som endrer membranpotensialet. Dette disponerer for maligne arytmier og danner grunnlaget for iskemiske EKG-forandringer.
Denne tilstanden er reversibel i en begrenset periode. Kardiomyocyttene kan tåle omtrent 20 til 30 minutter med alvorlig iskemi før irreversible skader (nekrose) inntreffer. Dersom reperfusjon ikke oppnås innen dette tidsvinduet, vil cellemembranene kollapse, enzymer (som troponiner) lekker ut, og cellen dør.
Den iskemiske kaskade
Forståelsen av myokardiskemi er ufullstendig uten konseptet om «den iskemiske kaskade». Dette beskriver den tidsmessige rekkefølgen av hendelser som oppstår når blodtilførselen reduseres:
- Perfusjonsdefekt: Redusert blodstrøm er den utløsende faktoren.
- Metabolsk endring: Overgang til anaerob metabolisme og laktatproduksjon.
- Diastolisk dysfunksjon: ATP-mangel hindrer rask reopptak av kalsium i sarkoplasmatisk retikulum, noe som fører til nedsatt relaksasjon (stiv ventrikkel).
- Systolisk dysfunksjon: Kontraksjonskraften reduseres (regional veggbevegelsesforstyrrelse).
- EKG-forandringer: ST-segmentendringer og T-bølgeinversjoner oppstår som følge av endrede elektriske egenskaper.
- Angina pectoris: Brystsmerter er ofte det siste symptomet i denne kaskaden.
Dette innebærer at en pasient kan ha betydelig iskemi med påvisbare veggbevegelsesforstyrrelser (f.eks. ved stressekkokardiografi) uten at EKG-forandringer eller smerter ennå har manifestert seg.
Myokardiskemi i klinisk praksis: koronarsykdom
I klinisk praksis skiller vi fundamentalt mellom «demand-iskemi» (økt behov) og «supply-iskemi» (redusert tilførsel). Ved stabil angina pectoris foreligger det et stabilt aterosklerotisk plakk som mekanisk hindrer blodstrømmen. I hvile er perfusjonen tilstrekkelig, men ved fysisk eller emosjonelt stress klarer ikke koronarkaret å dilatere ytterligere for å møte det økte oksygenkravet. Dette resulterer i reversibel iskemi som forsvinner ved hvile eller nitrater.
Stabil angina pectoris diagnostiseres ofte ved arbeidsbelastningstester (arbeids-EKG, stress-ekko eller stress-MR). Hensikten er å fremprovosere iskemi under kontrollerte former. Ettersom det subendokardiale laget (innerst mot ventrikkelen) er mest sårbart for iskemi på grunn av høyest veggstress og lavest perfusjonstrykk, vil iskemien typisk starte her.
Ved akutt koronarsyndrom (ACS) er patofysiologien annerledes. Her skyldes iskemien en plutselig reduksjon i oksygentilførsel («supply-iskemi») forårsaket av plakkruptur eller plakkerosjon med påfølgende trombose. Dette gir symptomer i hvile. Graden av okklusjon avgjør om det utvikler seg til transmural iskemi (som gir ST-elevasjon, STEMI) eller subendokardiell iskemi (som gir ST-depresjon/T-inversjon, NSTEMI/UAP).
Tabell 1: Myokard- og EKG-reaksjon i ulike situasjoner med iskemi eller økt arbeidsbelastning
| STATUS AV KORONARARTERIE | SITUASJON | EFFEKT | EKG REAKSJON |
| Normal koronararterie (ingen aterosklerose) | I hvile | Oksygentilførselen er tilstrekkelig, og iskemi kan ikke oppstå. | Ingen endringer på hvile-EKG. |
| Økt myokardial arbeidsbelastning (trening) | Oksygentilførselen øker parallelt med økt oksygenforbruk under trening. Dermed er tilførsel og etterspørsel balansert, og det oppstår ingen iskemi. | Ingen forandringer på hvile-EKG.
(Merk: Noen friske personer kan vise fysiologisk ST-depresjon med raskt oppadstigende J-punkt under maksimal belastning; dette er ikke tegn på iskemi). |
|
| Stabil koronarsykdom (aterosklerotisk plakk som forårsaker >70 % stenose) | I hvile | Koronar autoregulering opprettholder hvileperfusjon distalt for stenosen. Ingen symptomer. | Ingen endringer på hvile-EKG. |
| Økt myokardial arbeidsbelastning (trening) | Oksygenbehovet øker, men stenosen begrenser flow-reserven. Resultatet er subendokardiell iskemi (demand-iskemi) og brystsmerter som er reversible. | Arbeids-EKG kan avdekke horisontale eller nedadgående ST-segmentsenkninger og T-bølgeinversjoner. ST-elevasjoner er svært sjeldne ved stabil angina (unntatt ved vasospasme). | |
| Stabil, men kritisk koronarsykdom (stenose >90 %) | I hvile | Koronar flow-reserve er oppbrukt selv i hvile. Pasienten kan ha symptomer ved minimal anstrengelse eller i hvile. | Hvile-EKG kan vise permanente ST-deviasjoner og/eller T-bølgeforandringer, evt. tegn på venstre ventrikkelhypertrofi eller tidligere infarkt. |
| Plakkruptur/erosjon (Akutt Koronarsyndrom) | Når som helst (uavhengig av aktivitet) | Total okklusjon: Gir transmural iskemi og akutt STEMI. Subtotal okklusjon: Gir subendokardiell iskemi og NSTEMI/UAP. Symptomer lindres lite av hvile. |
STEMI: ST-elevasjon (skadestrøm). NSTEMI/UAP: ST-depresjon, T-bølgeinversjon, eller normalt EKG initialt. |
| Variantangina (Prinzmetals angina) | Vasospasme i koronararterien | Forbigående total okklusjon forårsaket av spasme i epikardial arterie, ofte uten betydelig aterosklerose. Gir transmural iskemi i hvile, ofte om natten. | Under anfall: Transiente ST-elevasjoner som normaliseres når smerten forsvinner. |
Tiden er muskel: Fra myokardiskemi til infarkt
Elektrofysiologisk grunnlag for EKG-forandringer
For å forstå hvorfor EKG-et endrer seg som det gjør, må vi se på cellemembranen. Ved iskemi svikter ATP-avhengige ionepumper. Dette fører til en utstrømming av kalium (K+) fra cellene og en delvis depolarisering av cellemembranen i hvile. Det iskemiske området får dermed et mindre negativt hvilemembranpotensial enn det friske vevet.
Denne forskjellen i potensial skaper en elektrisk strøm – en «skadestrøm» (current of injury) – mellom det iskemiske og det friske vevet.
- Ved transmural iskemi (STEMI): Skadestrømmen resulterer i en vektoriell forskyvning som løfter ST-segmentet i avledninger som ser mot det skadde området.
- Ved subendokardiell iskemi (NSTEMI/Angina): Skadestrømmen peker bort fra elektrodene på overflaten, noe som resulterer i ST-depresjon.
Varigheten av iskemien er avgjørende for utfallet. Myokard som forsynes av den okkluderte arterien, blir umiddelbart iskemisk og slutter å trekke seg sammen («stunned myocardium» i akuttfasen). Som nevnt ovenfor går cellene over til anaerob metabolisme for å opprettholde basal celleintegritet. Dette gjør at cellene kan overleve 20-30 minutter med alvorlig iskemi. Dette tidsvinduet er kritisk i akuttbehandling.
Hvis koronarstrømmen ikke gjenopprettes, vil celledød (nekrose) inntreffe. Utbredelsen av nekrosen følger et spesifikt mønster beskrevet som Reimer og Jennings’ bølgefrontfenomen. Nekrosen starter alltid i det mest sårbare området, som er subendokardiet, og sprer seg deretter som en bølgefront utover mot epikardiet over tid. Se figur 2.
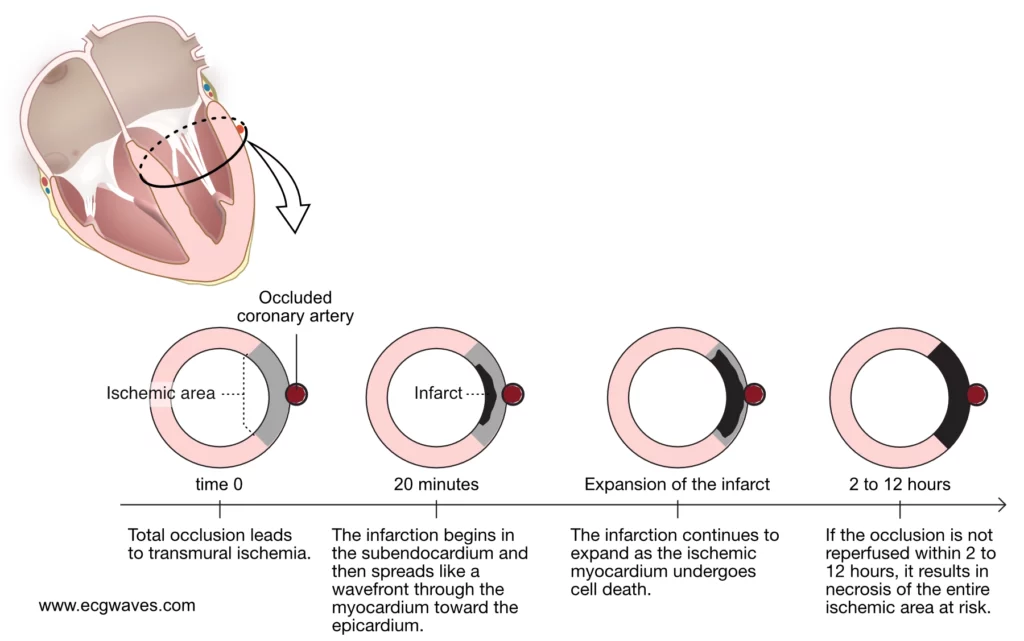
Tiden det tar før alt iskemisk myokard blir infarkert (fullført infarkt), varierer betydelig mellom pasienter. I lærebøker er det tradisjonelt antydet at infarktet er komplett i løpet av 4 til 6 timer ved en permanent okklusjon. Nyere kliniske studier og MR-studier viser imidlertid at prosessen kan være mer dynamisk, og tidsvinduet for myokardredning kan strekke seg fra 2 opp til 12 timer etter symptomdebut.
Flere faktorer påvirker hastigheten på infarktutviklingen:
- Kollateral sirkulasjon: Pasienter med langvarig koronarsykdom har ofte utviklet små omkjøringsveier (kollateraler) fra andre koronararterier. Disse kan opprettholde en viss perfusjon til det iskemiske området («rest-flow») og dermed forsinke celledøden betydelig.
- Iskemisk prekondisjonering: Korte episoder med iskemi (angina) før selve infarktet kan aktivere intracellulære beskyttelsesmekanismer som gjør cellene mer motstandsdyktige mot langvarig oksygenmangel.
- Myokardialt oksygenforbruk: Høy hjertefrekvens og høyt blodtrykk under infarktet akselererer nekrosen.
Totale okklusjoner (som resulterer i akutt STEMI) medfører transmural nekrose med mindre reperfusjonsbehandling (PCI eller trombolyse) iverksettes raskt. Selv om omtrent en tredjedel av okklusjoner rekanaliseres spontant innen 12–24 timer på grunn av kroppens eget fibrinolytiske system, er dette ofte for sent til å redde myokard. Likevel er sen åpning av arterien gunstig for sårtilheling og ventrikulær remodellering.
Forskjellen på «Stunned» og «Hibernating» myokard
Det er viktig å skille mellom to tilstander av levende, men dysfunksjonelt myokard, da revaskularisering har ulik effekt:
- Stunned myocardium («svimeslått» myokard): Dette oppstår etter en akutt, forbigående iskemisk episode (f.eks. etter vellykket PCI). Vevet er reperfundert og levedyktig, men kontraktiliteten er nedsatt i dager til uker før den normaliseres spontant.
- Hibernating myocardium («dvalemyokard»): Dette er en tilstand ved kronisk, alvorlig iskemi. For å overleve den reduserte blodstrømmen nedregulerer cellene sin metabolisme og slutter å trekke seg sammen. Ved «hibernation» vil funksjonen kun bedres dersom man gjenoppretter blodstrømmen (ved PCI eller CABG). Dette er klinisk viktig ved utredning av hjertesvikt med koronarsykdom.
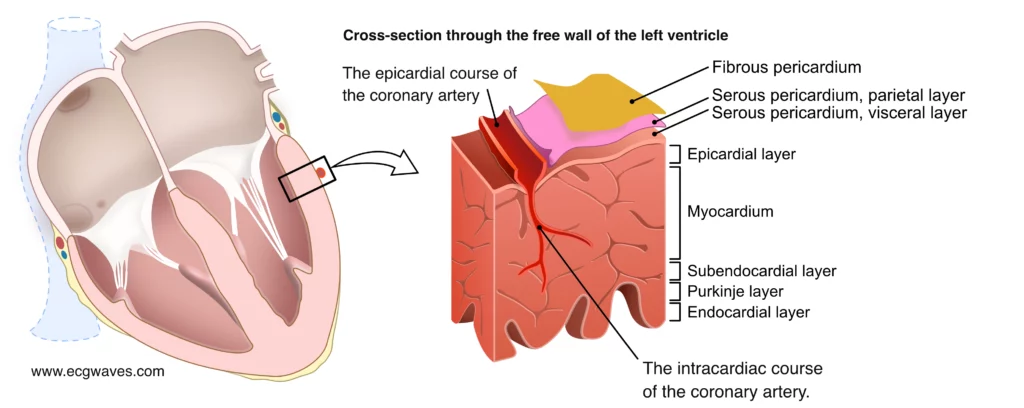
Figur 3 illustrerer anatomien bak sårbarheten i subendokardiet. Koronararteriene ligger på utsiden av hjertet (epikardiet) og sender perforerende grener inn gjennom muskelveggen. Blodstrømmen til subendokardiet må passere gjennom hele veggtykkelsen. Under systolen (sammentrekningen) komprimeres disse små karene av det høye trykket i venstre ventrikkel, noe som betyr at subendokardiet primært perfunderes i diastolen. Dette, kombinert med at subendokardiet har det høyeste oksygenforbruket på grunn av høyt veggstress, forklarer hvorfor iskemisk skade alltid starter her og sprer seg utover.