Forløpet fra plutselig hjertestans (SCA) til død følger en dynamisk patofysiologisk prosess som i stor grad påvirker effektiviteten av medisinske intervensjoner. Omfattende forskning viser at de tilgjengelige tiltakene er sterkt tidssensitive. For å maksimere sannsynligheten for gjenopprettet spontansirkulasjon (ROSC) og intakt nevrologisk overlevelse, bør tiltakene ideelt sett tilpasses den spesifikke fasen pasienten befinner seg i. Gjeldende internasjonale retningslinjer (som ERC og NRR) generaliserer imidlertid behandlingen, primært grunnet manglende metoder for å sanntidsbestemme hvilken metabolsk fase pasienten er i prehospitalt (utover anbefalingen om umiddelbar defibrillering ved bevitnet hjertestans med sjokkbar rytme).
De tre fasene etter en plutselig hjertestans ble først systematisert av Weisfeldt og Becker (2002). Basert på grunnleggende fysiologiske prinsipper, samt omfattende observasjonsstudier og dyreeksperimentelle modeller, beskrev de tidsvinduer som krever distinkte behandlingsstrategier for å optimalisere overlevelsen (Tabell 1).
| Fase | Periode | Optimal behandling |
|---|---|---|
| Elektrisk fase | 0 til 4 minutter | Umiddelbar defibrillering |
| Sirkulasjonsfase | 4 til 10 minutter | Kompresjoner og ventilasjon (priming), etterfulgt av defibrillering |
| Metabolsk fase | >10 minutter | Avanserte tiltak (eCPR/ECMO), farmakologi, hypotermi (historisk: «ukjent») |
Eksistensen av disse fasene underbygges av en lang rekke eksperimentelle og kliniske data. Den elektriske fasen og sirkulasjonsfasen forklares primært av elektrofysiologiske og energimessige endringer i myokardiet. Den siste fasen, den metabolske fasen, er kompleks og multifaktoriell. Den domineres av konsekvensene av global iskemi, intracellulær acidose, elektrolyttforstyrrelser (spesielt kalsiumoverskudd), inflammasjonskaskader og reperfusjonsskade ved gjeninnsetting av oksygenering.
Myokardiets oksygenbehov og energimetabolisme
Hjertet er et obligat aerobt organ, hvilket betyr at myokardiet er avhengig av kontinuerlig oksygentilførsel for oksidativ fosforylering og ATP-syntese. ATP er den universelle energikilden som driver både den mekaniske kontraksjonen og de energikrevende ionepumpene (Na+/K+-ATPase og Ca2+-ATPase) som opprettholder membranpotensialet.
Hjertet har en enorm metabolsk omsetning og forbruker omtrent 30 kilo ATP per dag (Neubauer et al.), noe som tilsvarer ca. 100 ganger hjertets egen vekt. Denne energiproduksjonen muliggjøres av en høy tetthet av mitokondrier, som utgjør nesten 30 % av kardiomyocyttenes volum (Page et al.). Under normale fysiologiske forhold kan mitokondriene regenerere hele ATP-lageret i løpet av sekunder (Fell et al., Houtkooper et al.). Mesteparten av ATP brukes til å drive det kontraktile maskineriet (aktin-myosin kryssbroer) og til å opprettholde ionemiljøet som er nødvendig for elektrisk stabilitet.
Ved sirkulasjonsstans opphører oksygentilførselen umiddelbart, og produksjonen av ATP stanser. Neumar og medarbeidere demonstrerte hvordan ATP-konsentrasjonen faller drastisk under ubehandlet ventrikkelflimmer (VF). Ved serielle myokardbiopsier fant de at ATP-lagrene tømmes raskt; etter kun 5 minutter med VF gjensto omtrent 50 % av utgangsnivået, og nivåene nærmet seg null etter 10–15 minutter.

Konsekvensene av ATP-tap er katastrofale for cellenes integritet. Uten ATP svikter ionepumpene, noe som fører til tap av membranpotensialet, akkumulering av intracellulært natrium og kalsium, samt cellulært ødem. Dette tapet av elektrokjemisk gradient gjør cellene mindre eksitable og svekker kontraktiliteten. Myokardnekrose og irreversibel celleskade («stone heart») inntreffer typisk innen 20–30 minutter etter total sirkulasjonskollaps.
Den elektriske fasen: Defibrillering
Det er estimert at en betydelig andel av hjertestans utenfor sykehus initieres av ventrikulær takykardi (VT) eller ventrikkelflimmer (VF). I den elektriske fasen (0–4 minutter) er myokardiet fortsatt relativt godt oksygenert og har tilstrekkelige ATP-lagre. Hjertet er dermed svært mottakelig for elektrisk konvertering.
Defibrillering er i denne fasen ekstremt effektivt. Dette bekreftes av data fra ICD-populasjoner (implanterbar kardioverterdefibrillator), hvor sjokk leveres nesten umiddelbart etter arytmidebut. ICD-er lykkes med å terminere VF/VT i opp mot 98 % av tilfellene (Zipes et al., Volosin et al.). Tilsvarende ser man ved overvåket hjertestans på sykehus (IHCA), hvor overlevelsen er ca. 70 % dersom sjokk gis innen 3 minutter (Hessulf et al.). Ved hjertestans utenfor sykehus (OHCA) faller overlevelsen dramatisk grunnet tiden det tar å nå pasienten. Hvis rytmen fortsatt er sjokkbar ved ankomst, er overlevelsen i gjennomsnitt 30 %, men dette varierer stort med responstid og kvalitet på lekmanns-HLR.
| Forsinkelse fra VT/VF-debut til defibrillering | Bevisgrunnlag | Suksessrate for defibrillering |
|---|---|---|
| <30 sekunder | ICD-studier | 98% |
| 3 minutter | Observasjonsstudier (IHCA) | 70% |
| <10-15 minutter | Observasjonsstudier (OHCA) | 20-30% |
For hvert minutt som går uten behandling, synker sannsynligheten for vellykket defibrillering med 7–10 %. Samtidig øker risikoen for at den kaotiske elektriske aktiviteten (VF) degenererer til asystoli (ingen elektrisk aktivitet). Figur 2 illustrerer fallet i overlevelse. Det er verdt å merke seg at tidlig oppstart av HLR («kjøpe tid») flater ut denne kurven noe, men erstatter ikke behovet for sjokk.
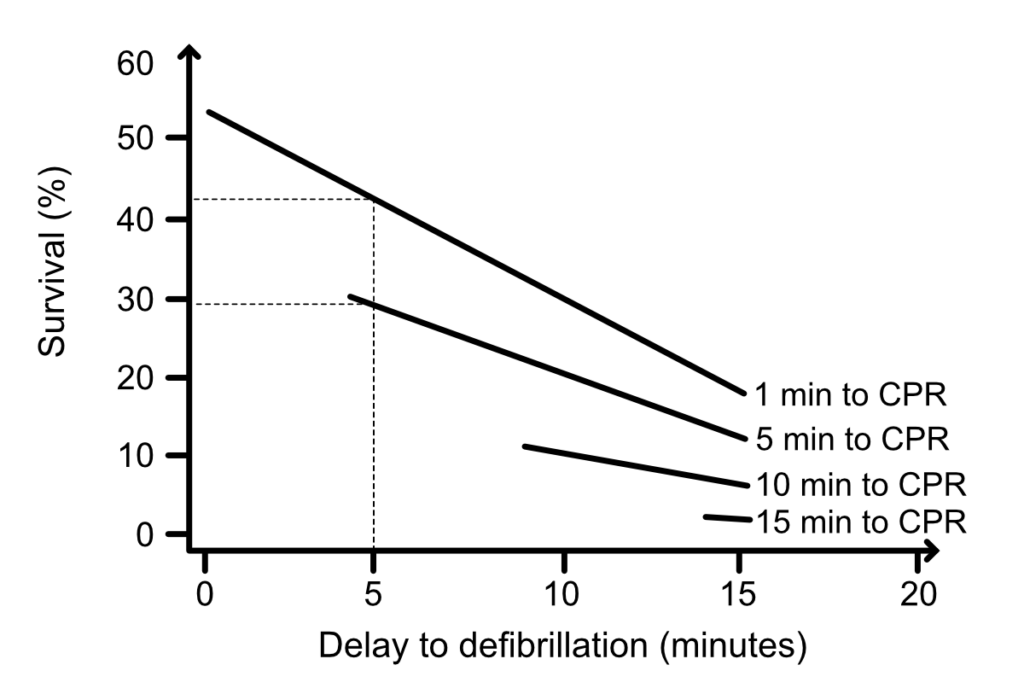
I den elektriske fasen (0–4 minutter) er sjokk førstevalg. Noen studier antyder at brystkompresjoner i denne helt initielle fasen kan ha liten tilleggsverdi eller til og med forsinke det livreddende sjokket (Niemann et al.). Derfor er umiddelbar bruk av hjertestarter (AED) eller manuell defibrillator prioritert.
Den sirkulatoriske fasen: Kompresjon og ventilasjon
Når hjertestansen har vedvart utover ca. 4 minutter, går pasienten inn i sirkulasjonsfasen (4–10 minutter). Her er myokardets energilagre redusert, og metabolitter (som laktat og CO2) har akkumulert seg i vevet. Myokardiet er nå iskemisk og «stivt», og sjansene for at et elektrisk sjokk alene skal konvertere rytmen til en perfunderende sinusrytme, er betydelig redusert.
Allerede i 1943 viste Gurvich et al. prinsippet om at defibrillering av langvarig VF krever forbehandling med brystkompresjoner. I denne fasen er formålet med brystkompresjoner ikke bare å levere oksygen til hjernen, men å generere et koronart perfusjonstrykk (CPP). Et tilstrekkelig CPP er nødvendig for å levere minimalt med oksygen til myokardcellene, vaske ut toksiske metabolitter og dermed «prime» hjertet for defibrillering.
Flere sentrale studier underbygger strategien om «kompresjoner før sjokk» i denne fasen:
- Yakaitis et al. viste at mens tidlig defibrillering var suverent best innen 3 minutter, falt suksessraten til 30 % etter 5 minutter. Dersom man imidlertid utførte kompresjoner før sjokket ved 5 minutter, økte suksessraten tilbake til 70 %.
- Niemann et al. demonstrerte at ved 7 minutters VF var defibrillering tre ganger mer effektiv etter en periode med HLR.
- Cobb et al. (Seattle-studien) analyserte OHCA-data og fant at ved responstider på over 4 minutter, ga 90 sekunder med HLR før første sjokk en 42 % relativ forbedring i overlevelse.
- En norsk studie av Wik et al. (Oslo) viste at 3 minutter med HLR før sjokk doblet sannsynligheten for ROSC ved responstider over 5 minutter.
Klinisk innebærer dette at kvaliteten på brystkompresjonene er avgjørende. Myokardiet blir ekstremt sensitivt for pauser («hands-off time»). Yu et al. viste at selv korte pauser (som pre-sjokk pauser for rytmeanalyse) kan være fatale. Etter en pause tar det tid å bygge opp koronart perfusjonstrykk igjen. Moderne retningslinjer fokuserer derfor sterkt på å minimere pauser («pre-shock pause» < 5 sekunder) og lade defibrillatoren mens kompresjoner pågår.

EKG-bildet er et direkte speilbilde av myokardiets metabolske tilstand. Parallelt med at ATP-nivåene faller, reduseres amplituden på ventrikkelflimmeret. Det går fra «grov VF» (som har høy sannsynlighet for sjokk-suksess) til «fin VF» (som sjelden responderer på sjokk). Behandling av fin VF med sjokk resulterer ofte i asystoli. Målet med kompresjoner i sirkulasjonsfasen er å forbedre metabolsk status slik at fin VF blir til grov VF før sjokk gis.
Den metabolske fasen: Iskemi og reperfusjonsskade
Den metabolske fasen inntreffer typisk etter ca. 10 minutter med hjertestans. Dette er den mest kritiske fasen med høyest mortalitet. På dette stadiet har pasienten utviklet dyp global iskemi, systemisk acidose og betydelige elektrolyttforstyrrelser. Selv om hjertet fortsatt kan ha sporadisk elektrisk aktivitet (oftest PEA eller fin VF), er myokardiet ofte «stunned» og ute av stand til å opprettholde sirkulasjon selv om rytmen normaliseres.
Patofysiologisk kjennetegnes denne fasen av at cellemembranene bryter sammen. Dette tillater massiv influks av kalsium i cellene, noe som aktiverer proteaser og fosfolipaser som bryter ned cellestrukturer. I tillegg sirkulerer inflammatoriske cytokiner og endotoksiner (fra iskemisk tarm) i blodet.
Paradokset med reperfusjonsskade
Dersom man lykkes med å gjenopprette sirkulasjon (ROSC) eller starter effektiv HLR i denne fasen, oppstår et nytt problem: Reperfusjonsskade. Når oksygenrikt blod returnerer til vev som har vært iskemisk over tid, genereres store mengder frie radikaler (reaktive oksygenforbindelser, ROS). Mitokondriene, som er skadet av iskemien, klarer ikke å håndtere oksygenet normalt. Dette fører til oksidativt stress som kan utløse apoptose (programmert celledød) og nekrose. Det finnes data som tyder på at ukontrollert reperfusjon kan akselerere myokardnekrose (Vanden Hoek et al.). Dette er bakgrunnen for at man i post-resusciteringsbehandling er forsiktig med hyperoksi (for mye oksygen).
Kliniske implikasjoner og avansert behandling (eCPR)
Tradisjonell HLR og defibrillering har svært lav suksessrate i den metabolske fasen. Hjernen begynner å få irreversible skader, og hjertet er refraktært mot konvensjonell behandling (adrenalin/amiodarone/sjokk). Tidligere ble behandlingsalternativene her ansett som «ukjente» eller ikke-eksisterende.
I moderne kardiologi har imidlertid eCPR (extracorporeal CPR), altså bruk av hjerte-lunge-maskin (ECMO) under pågående hjertestans, dukket opp som en potensiell bro over den metabolske fasen. Ved å etablere mekanisk sirkulasjon kan man gjenopprette perfusjon til vitale organer, korrigere acidose og metabolske forstyrrelser, og gi hjertet tid til å hvile eller behandles (f.eks. ved akutt PCI). Dette krever imidlertid at pasienten selekteres strengt og at systemet er rigget for rask kanylering, ettersom tidsvinduet for å redde hjernen fortsatt er begrenset.
Referanser
Kaustubha D. Patil, Henry R. Halperin, Lance B. Becker. Gjenopplivning og reperfusjon ved hjertestans. Circulation Research (2015).
Neubauer S. Det sviktende hjertet – en motor uten drivstoff. New England Journal of Medicine. 2007;356:1140-1151
Fell DA, Sauro HM. Metabolsk kontrollanalyse. Effekten av høye enzymkonsentrasjoner. European Journal of Biochemistry. 1990;192:183-187
Houtkooper RH, Canto C, Wanders RJ, Auwerx J. Det hemmelige livet til nad: En gammel metabolitt som kontrollerer nye metabolske signalveier. Endokrine anmeldelser. 2010;31:194-223
Gurvich NL, Yuniev GS. Gjenoppretting av hjerterytmen under fibrillering ved hjelp av en kondensatorutladning. Am Rev Sov Med 1947;4:252-6.
Vanden Hoek TL, Shao Z, Li C, Zak R, Schumacker PT, Becker LB. Reperfusjonsskade i hjertemyocytter etter simulert iskemi. Am J Physiol. 1996;270: H1334-H1341. 30.
Vanden Hoek TL, Qin Y, Wojcik K, et al. Reperfusjon, ikke simulert iskemi, initierer intrinsisk apoptoseskade i kyllingkardiomyocytter. Am J Physiol Heart Circ Physiol.
Yakaitis RW, Ewy GA, Otto CW, Taren DL, Moon TE. Innflytelse av tid og terapi på ventrikulær defibrillering hos hunder. Crit Care Med. 1980;8:157-163.
Menegazzi JJ, Davis EA, Yealy DM, et al. En eksperimentell algoritme versus standard avansert hjertestøtte i en svinemodell av hjertestans utenfor sykehus. Ann Emerg Med. 1993;22:235-239. 23.
Menegazzi JJ, Seaberg DC, Yealy DM, Davis EA, MacLeod BA. Kombinasjon av farmakoterapi med forsinket motsjokk vs standard avansert hjertestøtte etter langvarig ventrikkelflimmer. Prehosp Emerg Care. 2000;4:31-37.
Niemann JT, Cairns CB, Sharma J, Lewis RJ. Behandling av langvarig ventrikkelflimmer. Circulation. 1992;85:281-287. 20.
Niemann JT, Cruz B, Garner D, Lewis RJ. Umiddelbart motsjokk versus hjerte-lungeredning før motsjokk i en 5-minutters svinemodell av ventrikkelflimmerarrest. Ann Emerg Med. 2000;36:543-546.
Garcia LA, Allan JJ, Kerber RE. Interaksjoner mellom HLR og defibrilleringskurver. Resuscitation. 2000;47:301-305.
Page E, McCallister LP. Kvantitativ elektronmikroskopisk beskrivelse av hjertemuskelceller. Anvendelse på normale, hypertrofierte og tyroksinstimulerte hjerter. Am J Cardiol. 1973;31:172-181.
Wik L, Hansen TB, Fylling F, et al. Delaying defibrillation to give basic cardiopulmonary resuscitation to patients with out-of-hospital ventricular fibrillation: a randomized trial. JAMA. 2003;289:1389-1395.